MetCCS Predictor is developed to predict CCS values for metabolites in ion mobility − mass spectrometry (IM-MS), and it allows everyone to predict CCS values of metabolites of interest. We find it can also be applicable for predicting of CCS values of other small chemical compounds like drugs, pesticides and so on. Users can simply import 14 common molecular descriptors of one metabolite to calculate its CCS values within seconds. The software employs a support vector regression (SVR) based machine-learning algorithm for prediction, and the general principle has been published on Analytical Chemistry (2016).1 We experimentally measured CCS values (ΩN2) of ∼400 metabolites in nitrogen buffer gas and used these values as training data to optimize the prediction method. Prediction precision of this method has been validated with a median relative error (MRE) of ~3%. Since CCS values of metabolites in the training data set were all acquired in nitrogen buffer gas, the predicted CCS values are all nitrogen CCS values.
In addition to prediction function, MetCCS Predictor also includes search and match functions. The database search function facilitates users to search CCS values of metabolites in MetCCS database using known HMDB ID, SMILES or InChI Identifier. Metabolite match function is designed for users to identify unknown metabolites using experimentally measured m/z and CCS values.
The workflow for MetCCS Prediction is divided into five steps: (1) import molecular descriptors, (2) check data quality, (3) impute missing values (if necessary), (4) predict CCS values, and (5) export results (Figure 1).

14 common molecular descriptors of one metabolite (or chemical compound) are used to predict its CCS values by MetCCS predictor. These descriptors and their suggested ranges are listed in the Table 1. All descriptors are calculated by cheminformatic software like ChemAxon and ALOGPS using the molecular structure. Human Metabolome Database (HMBD) also provides the values of molecular descriptors. Users can directly input molecular descriptors into the textbox or import as a CSV file.
| No. | Name | Definition | Source | Range |
|---|---|---|---|---|
| 1 | Exact_Mass | The exact mass of compound | Molecular formula | [100, 1000] |
| 2 | Formal_Charge | The formal charge | ChemAxon | [-2, 1] |
| 3 | Physiological_Charge | The physiological charge | ChemAxon | [-8, 4] |
| 4 | logP_ALOGPS | The octanol/water partition coefficient | ALOGPS | [-4, 10] |
| 5 | logP_ChemAxon | The octanol/water partition coefficient | ChemAxon | [-11, 12] |
| 6 | logS | The aqueous solubility | ALOGPS | [-8, 1] |
| 7 | pKa_Strongest_Acidic | The acid dissociation constant | ChemAxon | [-7, 20] |
| 8 | pKa_Strongest_Basic | The basic dissociation constant | ChemAxon | [-10, 12] |
| 9 | Hydrogen_Acceptor_Count | The sum of the acceptor atoms | ChemAxon | [0, 21] |
| 10 | Hydrogen_Donor_Count | The sum of the donor atoms | ChemAxon | [0, 14] |
| 11 | Polar_Surface_Area | The sum of the all polar atoms surface in a molecule | ChemAxon | [0, 400] |
| 12 | Rotatable_Bond_Count | The number of rotatable bonds in the molecule | ChemAxon | [0, 43] |
| 13 | Refractivity | Molar refractivity | ChemAxon | [0, 252] |
| 14 | Polarizability | Molecular polarizability | ChemAxon | [0, 105] |
The units of descriptors: Exact mass (Da); Polar Surface Area (Å2); Refractivity (m3·mol-1); Polarizability (Å3).
As shown in Figure 2a, the software first checks the quality of the imported molecular descriptors, including availability of exact mass and the number of missing values. "Exact mass" is the mandatorily required molecular descriptor for the prediction. Therefore, the value of "Exact_mass" must be imported, otherwise, MetCCS Predictor returns error information. The number of missing values (denoted as "NA") is also checked, and the maximum tolerance of missing values is set as 7. If the number of missing descriptors is less than 7, warning information is given out.
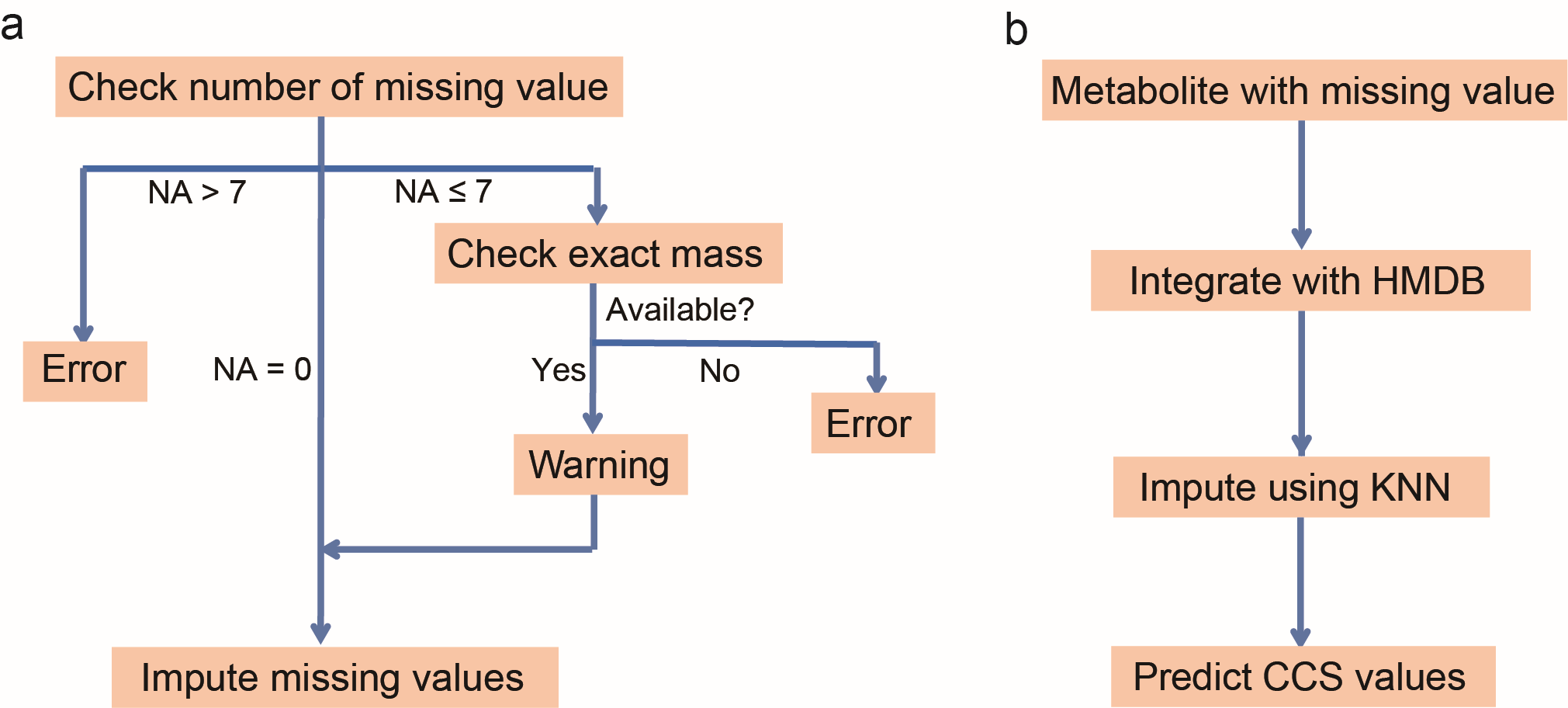
Before the prediction of CCS values, missing values for molecular descriptors are imputed using K-Nearest Neighbor algorithm (KNN).2 All imported descriptors are first integrated with dataset of HMDB (www.hmdb.ca), and 10 of the most similar metabolites in terms of molecular descriptors across all metabolites in HMDB are chosen. The weighted average of these 10 metabolites is calculated and replaces the corresponding missing value based on their similarity. Then, all descriptors are saved and transferred to the prediction model.
The method of CCS prediction was introduced in our previous publication. Briefly, MetCCS Predictor employs SVR algorithm to implicitly map molecular descriptors of metabolites into a high-dimensional feature space using a kernel function, and to construct a hyperplane in that space to perform the high-dimensional regression between molecular descriptors and CCS values in the training dataset. For more detailed information, please refer to our publication.
The prediction results of metabolites are listed in Table 2. For each metabolite, CCS values for 5 ion adducts are predicted, such as [M+H]+, [M+Na]+ and [M+H-H2O]+ in positive mode, and [M-H]- and [M+Na-2H]- in negative mode.
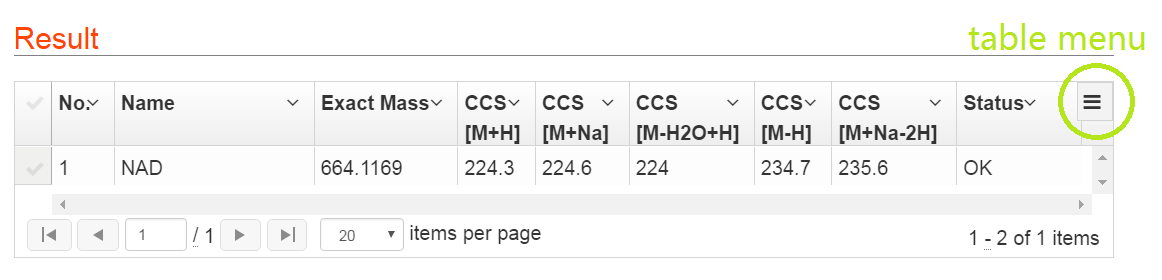
Explanations for Status:
Users can readily search CCS values of metabolites in the MetCCS database. This function allows users to directly search database in three different ways: HMDB ID, SMILES or InCHI identifiers. It also supports batch search function with a maximum of 100 query lines per request. The MetCCS database contains 35,203 metabolites with exact mass between 60 and 1000 Da, accounting for 176,015 CCS values for 5 different adducts. If a metabolite is not included in the database, a hint message will give out.
This function is designed for users to identify unknown metabolites using experimentally measured m/z and CCS values. Users are required to input both of the m/z and CCS values of an unknown metabolite and define a proper tolerance for the m/z and CCS value measurements together with polarity information. The web server will return metabolite candidates in the MetCCS database within the defined tolerances. For a metabolite with m/z 332.0746 Da and CCS value 168 Å2, we define the tolerance of m/z and CCS values as 15 ppm and 3% respectively. After metabolite match, this metabolite is identified as dAMP. The m/z accuracy is 4 ppm and CCS relative error is 1.3%. In addition, we also provide HMDB ID of this metabolite, and user can click the HMDB ID to link, and get more information about the metabolite in HMDB website. Users also can click grid menu to download match results as CSV file. The result of this example is shown in Figure 4.
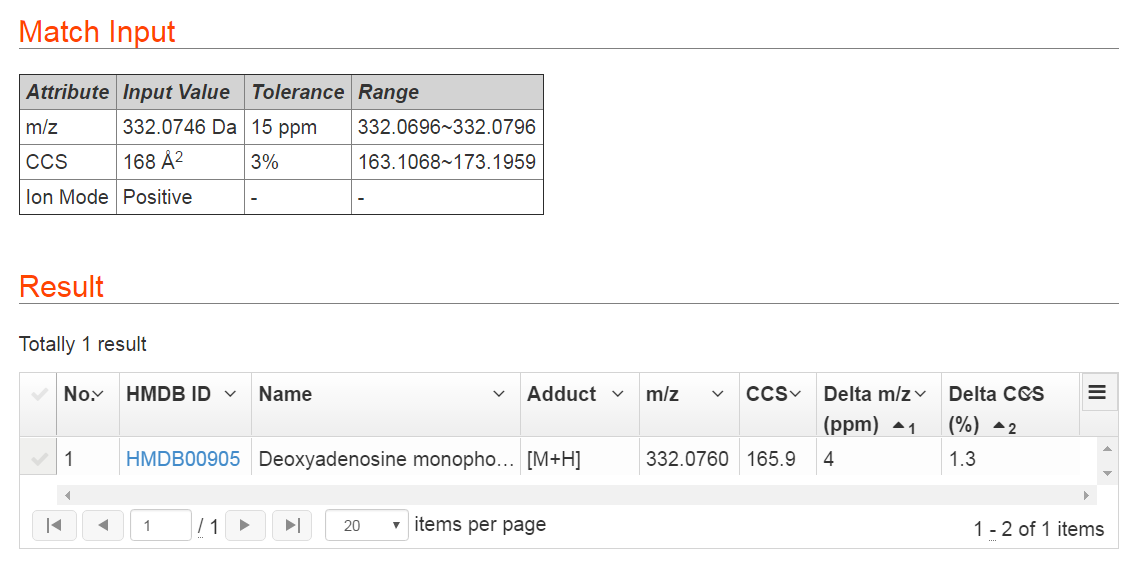
The equations to calculate delta m/z and delta CCS values are shown as Eq. 1 and 2.
Feb 17th, 2017: Added MetCCS Database Search and Metabolite Match
Nov 9th, 2016: Added batch mode
Oct 24th, 2016: Created this site
(1) Zhou, Z.; Shen, X.; Tu, J.; Zhu, Z. J. Anal. Chem. 2016, 88, 11084-11091.
(2) Hastie, T.; Tibshirani, R.; Narasimhan, B.; Chu, G.; R package.